





Resources
Read our latest press releases

Read our most recent press releases

Press Release
Loftware Unveils Enhanced Cloud Labeling Platform at LogiMAT 2024 - Offering Gateway to Intelligent and Transparent...
Loftware, the global leader in Enterprise Labeling and Artwork Management solutions, today unveiled Loftware Cloud at...

Press Release
Loftware Spectrum 5.0 Achieves SAP® Certified Integration with RISE with SAP S/4HANA® Cloud
Loftware, the largest global software company specializing in Enterprise Labeling and Artwork Management solutions,...
Press Release
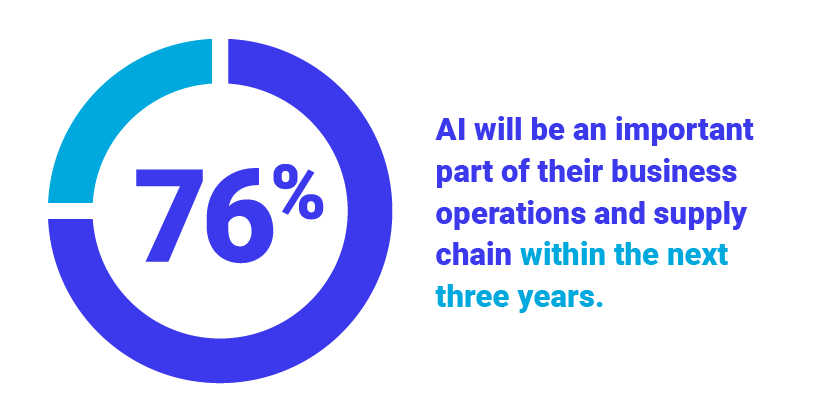
Global Survey Highlights Rise of Technology-Infused Supply Chains to Address Disruption, Uncertainty, and Cost Pressures
More than half (52%) of companies currently host critical enterprise applications in the Cloud while 76% believe...

Press Release
Loftware Recognized as One of New Hampshire’s "Top 100 Private Companies" for Seventh Year
Loftware, the global leader in Enterprise Labeling and Artwork Management solutions, today announced that it has been...